Distrofia miotonica (DM)
Scarica la nostra scheda informativa sulla distrofia miotonica (DM)
Che cos’è la distrofia miotonica (DM)?
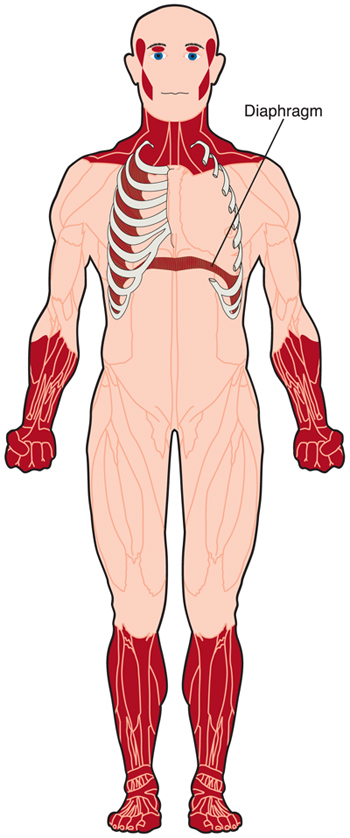 La distrofia miotonica (DM) è una forma di distrofia muscolare che colpisce i muscoli e molti altri organi del corpo.
La distrofia miotonica (DM) è una forma di distrofia muscolare che colpisce i muscoli e molti altri organi del corpo.
La parola “miotonica” è la forma aggettivale della parola “miotonia”, definita come incapacità di rilassare i muscoli a volontà.
Il termine “distrofia muscolare” significa degenerazione muscolare progressiva, con debolezza e restringimento del tessuto muscolare.,
La distrofia miotonica è spesso abbreviata come ” DM ” in riferimento al suo nome greco, dystrophia myotonica. Un altro nome usato occasionalmente per questo disturbo è la malattia di Steinert, dopo il medico tedesco che originariamente descrisse il disturbo nel 1909.
Quali sono le cause DM?
DM è diviso in due tipi.
Il DM di tipo 1 (DM1), noto da tempo come malattia di Steinert, si verifica quando un gene sul cromosoma 19 chiamato DMPK contiene una sezione anormalmente espansa situata vicino alla regione di regolazione di un altro gene, SIX5.,
Il tipo 2 DM (DM2), riconosciuto nel 1994 come una versione più mite di DM1, è causato da una sezione anormalmente espansa in un gene sul cromosoma 3 chiamato ZNF9. DM2 è stato originariamente chiamato PROMM, per miopatia miotonica prossimale, un termine che è rimasto in uso ma è un po ‘ meno comune del termine DM2.
Le sezioni espanse del DNA in questi due geni sembrano avere molti effetti complessi su vari processi cellulari. Sia in DM1 che in DM2, l’espansione ripetuta viene trascritta in RNA ma rimane non tradotta in proteine.,
Queste condizioni sono alcune delle forme più comuni di distrofia muscolare ad esordio adulto. DM è la distrofia muscolare più comune tra gli adulti di origine europea. La prevalenza di DM è di circa 10 casi per 100.000 individui.1,2,3,4 Tra le popolazioni non bianche, DM1 è raro o raro.5,6,7,8 Rapporti dall’Europa suggeriscono che la prevalenza di DM2 è simile a quella di DM1.
Quali sono i sintomi di DM?,
DM causa debolezza dei muscoli volontari, anche se il grado di debolezza e i muscoli più colpiti variano notevolmente a seconda del tipo di DM e dell’età della persona con il disturbo.
La miotonia, l’incapacità di rilassare i muscoli a volontà, è un’altra caratteristica di DM. Ad esempio, potrebbe essere difficile per qualcuno con DM lasciare andare la mano di qualcuno dopo averla agitata.
Con il progredire della malattia, il cuore può sviluppare un ritmo anormale e il muscolo cardiaco può indebolirsi. I muscoli utilizzati per la respirazione possono indebolirsi, causando una respirazione inadeguata, in particolare durante il sonno.,9
Inoltre, nel DM di tipo 1, i muscoli involontari, come quelli del tratto gastrointestinale, possono essere influenzati. Possono verificarsi difficoltà di deglutizione, stitichezza e calcoli biliari.10,11 Nelle femmine, i muscoli dell’utero possono comportarsi in modo anomalo, portando a complicazioni durante la gravidanza e il travaglio.12,13
Lo sviluppo della cataratta (macchie opache nelle lenti degli occhi) relativamente presto nella vita è un’altra caratteristica del DM, sia nel tipo 1 che nel tipo 2.,14
L’intelligenza generale è di solito normale nelle persone con DM, ma le difficoltà di apprendimento e un comportamento apatico sono comuni nella forma di tipo 1.15 Nel DM1 congenito, che colpisce i bambini dal momento della nascita, può esserci una grave compromissione del funzionamento cognitivo. Questi bambini possono anche avere problemi di parola,udito, 16 e affaticamento della vista.
Generalmente, prima inizia DM1, più profondi tendono ad essere i sintomi. Per ulteriori informazioni, vedere Segni e sintomi.
DM2 ha una prognosi complessiva migliore di DM1. I sintomi sono spesso relativamente lievi e progrediscono lentamente., DM2 si verifica raramente durante l’infanzia e non esiste una forma congenita nota di DM2.
Qual è la progressione del DM?
La progressione della DM varia notevolmente tra gli individui, ma in generale i sintomi progrediscono gradualmente. L’aspettativa di vita è chiaramente ridotta per i pazienti con DM1 congenito ed è probabilmente ridotta per i pazienti con DM1 infantile e DM1 classico (ad esordio adulto).
Il tipo più comune di DM1 — la forma ad esordio adulto-inizia nell’adolescenza o nella giovane età adulta, spesso con debolezza nei muscoli del viso, del collo, delle dita e delle caviglie., La debolezza è lentamente progressiva per questi e, infine, altri muscoli.
Quando DM1 inizia prima dell’adolescenza — le forme di insorgenza congenita e di insorgenza infantile della malattia-può essere molto diverso nella progressione dal tipo di esordio adulto. I bambini con DM1 ad esordio congenito, una volta che sopravvivono al periodo neonatale cruciale della debolezza muscolare respiratoria con l’aiuto della ventilazione assistita, di solito mostrano miglioramenti nelle funzioni motorie e respiratorie. Possono avere deficit cognitivo, ritardo del linguaggio, difficoltà a mangiare e bere e vari altri ritardi nello sviluppo.,15
La forma di esordio infantile di DM1, prima dei 10 anni, è più spesso caratterizzata da anomalie cognitive e comportamentali che da disabilità fisiche, come compromissione intellettuale, deficit di attenzione, disfunzione esecutiva, ansia e disturbi dell’umore.17, 18, 19 Alla fine, i sintomi muscolari si sviluppano, a vari livelli.
DM2 è, in generale, una malattia più lieve rispetto al tipo 1. Non sembra avere una forma a esordio congenito e raramente inizia nell’infanzia.,
In contrasto con il tipo 1 DM, i muscoli colpiti prima in DM2 sono i muscoli prossimali — quelli vicino al centro del corpo — in particolare intorno ai fianchi. Tuttavia, qualche debolezza dito può essere visto presto pure. Il disturbo progredisce lentamente, ma la mobilità può essere compromessa precocemente a causa della debolezza dei muscoli grandi e portanti.
Qual è lo stato della ricerca su DM?
L’identificazione delle mutazioni genetiche alla base di DM1 e DM2 e la comprensione almeno in parte di come le mutazioni causano la malattia, ha aperto strade per lo sviluppo della terapia in DM.,
La maggior parte delle strategie attualmente in fase di sviluppo mirano a bloccare gli effetti dannosi del DNA espanso nel gene DMPK (tipo 1) o nel gene ZNF9 (tipo 2). Per di più, vedere la ricerca, A fuoco: Distrofia miotonica, e in particolare DM Ricerca: Cercando di liberare le proteine da un ‘ragnatela tossica.’