distrofia miotonică (dm)
descărcați fișa informativă a distrofiei miotonice (dm)
ce este distrofia miotonică (DM)?
 distrofiei Miotonice (DM) este o formă de distrofie musculară care afectează mușchii și multe alte organe din corp.cuvântul ” miotonic „este forma adjectivală a cuvântului” miotonie”, definită ca o incapacitate de relaxare a mușchilor la alegere.termenul „distrofie musculară” înseamnă degenerare musculară progresivă, cu slăbiciune și contracție a țesutului muscular.,distrofia miotonică este adesea abreviată ca ” DM ” cu referire la numele său grecesc, dystrophia myotonica. Un alt nume folosit ocazional pentru această tulburare este boala Steinert, după medicul German care a descris inițial tulburarea în 1909.
distrofiei Miotonice (DM) este o formă de distrofie musculară care afectează mușchii și multe alte organe din corp.cuvântul ” miotonic „este forma adjectivală a cuvântului” miotonie”, definită ca o incapacitate de relaxare a mușchilor la alegere.termenul „distrofie musculară” înseamnă degenerare musculară progresivă, cu slăbiciune și contracție a țesutului muscular.,distrofia miotonică este adesea abreviată ca ” DM ” cu referire la numele său grecesc, dystrophia myotonica. Un alt nume folosit ocazional pentru această tulburare este boala Steinert, după medicul German care a descris inițial tulburarea în 1909.
ce cauzează DM?
DM este împărțit în două tipuri.
Tip 1 DZ (dz de tip 1), cunoscut mult timp ca boala Steinert, apare atunci când o genă de pe cromozomul 19 numit DMPK conține un anormal extins secțiunea situat aproape de regulament regiune de o altă genă, SIX5.,
tipul 2 DM (DM2), recunoscut în 1994 ca o versiune mai blândă a DM1, este cauzat de o secțiune anormal de extinsă într-o genă de pe cromozomul 3 numită ZNF9. DM2 a fost inițial numit PROMM, pentru miopatie miotonică proximală, un termen care a rămas în uz, dar este ceva mai puțin obișnuit decât termenul DM2.secțiunile extinse ale ADN-ului din aceste două gene par să aibă multe efecte complexe asupra diferitelor procese celulare. Atât în DM1, cât și în DM2, expansiunea repetată este transcrisă în ARN, dar rămâne netradusă în proteine.,aceste afecțiuni sunt unele dintre cele mai frecvente forme de distrofie musculară cu debut adult. DM este cea mai frecventă distrofie musculară în rândul adulților de origine europeană. Prevalența DM este de aproximativ 10 cazuri la 100.000 de persoane.1,2,3,4 dintre populațiile nonwhite, DM1 este mai puțin frecventă sau rară.5,6,7,8 rapoarte din Europa sugerează că prevalența DM2 este similară cu cea a DM1.
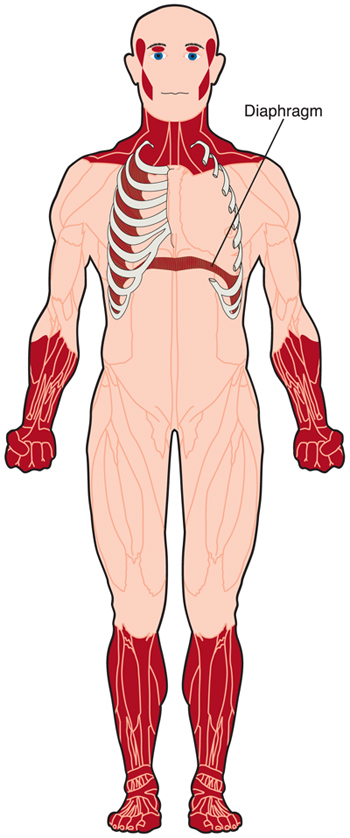
care sunt simptomele DM?,
DM cauzează slăbiciunea mușchilor voluntari, deși gradul de slăbiciune și mușchii cei mai afectați variază foarte mult în funcție de tipul de DM și de vârsta persoanei cu tulburare.miotonia, incapacitatea de a relaxa mușchii la voință, este o altă caracteristică a DM. De exemplu, poate fi dificil pentru cineva cu DM să renunțe la mâna cuiva după ce a scuturat-o.pe măsură ce boala progresează, inima poate dezvolta un ritm anormal, iar mușchiul inimii poate slăbi. Mușchii utilizați pentru respirație pot slăbi, provocând o respirație inadecvată, în special în timpul somnului.,9
În plus, în tipul 1 DM, mușchii involuntari, cum ar fi cei ai tractului gastro-intestinal, pot fi afectați. Pot apărea dificultăți la înghițire, constipație și calculi biliari.10,11 la femele, mușchii uterului se pot comporta anormal, ducând la complicații în timpul sarcinii și al travaliului.12,13
dezvoltarea cataractei (pete opace în lentilele ochilor) relativ devreme în viață este o altă caracteristică a DM, atât în tipul 1, cât și în tipul 2.,14
inteligența generală este de obicei normală la persoanele cu DM, dar dizabilități de învățare și un comportament apatic sunt comune în forma de tip 1.15 în DM1 congenital, care afectează copiii din momentul nașterii, poate exista o afectare gravă a funcționării cognitive. Acești copii pot avea, de asemenea,probleme cu vorbirea, auzul, 16 și oboseala vederii.în general, cu cât DM1 începe mai devreme, cu atât simptomele tind să fie mai profunde. Pentru mai multe informații, consultați semne și simptome.
DM2 are un prognostic general mai bun decât DM1. Simptomele sunt adesea relativ ușoare și progresează lent., DM2 apare rar în timpul copilăriei și nu există o formă cunoscută de debut congenital de DM2.
care este progresia DM?progresia DM variază foarte mult între indivizi, dar, în general, simptomele progresează treptat. Speranța de viață este în mod clar redusă pentru pacienții cu DM1 congenital și este probabil redusă pentru pacienții cu dm1 din copilărie și DM1 clasic (cu debut adult).cel mai frecvent tip de DM1 — forma cu debut adult-începe în adolescență sau la vârsta adultă, adesea cu slăbiciune a mușchilor feței, gâtului, degetelor și gleznelor., Slăbiciunea este lent progresivă pentru acești și, eventual, pentru alți mușchi.când DM1 începe mai devreme în viață decât adolescența – formele cu debut congenital și cu debut în copilărie ale bolii-poate fi destul de diferită în progresie față de tipul cu debut adult. Copiii cu DM1 cu debut congenital, odată ce supraviețuiesc perioadei neonatale cruciale de slăbiciune musculară respiratorie cu ajutorul ventilației asistate, prezintă de obicei îmbunătățiri ale funcțiilor motorii și respiratorii. Acestea pot avea tulburări cognitive, vorbire întârziată, dificultăți în a mânca și a bea și diverse alte întârzieri de dezvoltare.,15
forma de debut a copilăriei DM1, înainte de vârsta de 10 ani, este mai des caracterizată de anomalii cognitive și comportamentale decât de dizabilități fizice, cum ar fi tulburări intelectuale, deficite atenționale, disfuncții executive, anxietate și tulburări de dispoziție.17, 18, 19 în cele din urmă, simptomele musculare se dezvoltă, în grade diferite.DM2 este, în general, o boală mai ușoară decât tipul 1. Nu pare să aibă o formă cu debut congenital și rareori începe în copilărie.,spre deosebire de tipul 1 DM, mușchii afectați mai întâi în DM2 sunt mușchii proximali — cei apropiați de centrul corpului — în special în jurul șoldurilor. Cu toate acestea, o anumită slăbiciune a degetului poate fi văzută și mai devreme. Tulburarea progresează încet, dar mobilitatea poate fi afectată devreme din cauza slăbiciunii mușchilor mari, purtători de greutate.