Podstawienie Nukleofilowe grupy hydroksylowej
chemiczne zachowanie halogenków alkilowych może być wykorzystane jako punkt odniesienia w odkrywaniu analogicznych reakcji podstawienia i eliminacji alkoholi. Główną różnicą jest oczywiście zmiana anionu z halogenku na wodorotlenek. Ponieważ tlen jest nieco bardziej elektronowy niż chlor (3,5 vs. 2,8 w skali Paulinga), oczekuje się, że Wiązanie C-O będzie bardziej polarne niż Wiązanie C-Cl., Ponadto niezależna miara elektrofilowych właściwości atomów węgla z ich przesunięć chemicznych NMR (zarówno protony 13C, jak i Alfa) wskazuje, że podstawniki tlenu i chloru wywierają podobny wpływ na wycofanie elektronów, gdy są połączone z hybrydyzowanymi atomami węgla sp3. Pomimo tych obiecujących dowodów, alkohole nie przechodzą tych samych reakcji SN2, które są powszechnie obserwowane z halogenkami alkilowymi. Na przykład, szybka reakcja SN2 1-bromobutanu z cyjankiem sodu, pokazana poniżej, nie ma równoległości, gdy 1-butanol jest traktowany z cyjankiem sodu., W rzeczywistości alkohol etylowy jest często stosowany jako rozpuszczalnik do reakcji substytucji halogenków alkilowych, takich jak ten.
CH3CH2CH2CH2–Br + NA(+) CN(–)  CH3CH2CH2CH2–CN + Na(+) Br(–)
CH3CH2CH2CH2–CN + Na(+) Br(–)
CH3CH2CH2CH2–OH + NA(+) CN(–) brak reakcji
kluczowym czynnikiem jest tutaj stabilność anionu pozostawiając (bromek vs wodorotlenek)., HBr jest dużo silniejszym kwasem niż woda (o ponad 18 rzędów wielkości), a różnica ta znajduje odzwierciedlenie w reakcjach, które generują odpowiednie sprzężone Zasady. Słabsza zasada, bromek, jest bardziej stabilna, a jej uwolnienie w reakcji substytucji lub eliminacji jest znacznie korzystniejsze niż jon wodorotlenkowy, mocniejsza i mniej stabilna zasada.
wyraźnym krokiem w kierunku poprawy reaktywności alkoholi w reakcjach SN2 byłoby zmodyfikowanie grupy funkcyjnej –OH w sposób, który poprawia jej stabilność jako anionu opuszczającego., Jedną z takich modyfikacji jest przeprowadzenie reakcji substytucji w mocnym kwasie, przekształcającej-OH do –OH2 (+). Ponieważ jon hydroniowy (H3O (+)) jest dużo silniejszym kwasem niż woda, jego sprzężona zasada (H2O) jest lepszą grupą wyjściową niż jon wodorotlenkowy. Jedynym problemem tej strategii jest to, że wiele nukleofilów, w tym cyjanek, jest dezaktywowanych przez protonację w silnych kwasach, skutecznie usuwając nukleofilowy Ko-reagent wymagany do substytucji., Silne kwasy HCl, HBr i HI nie podlegają tej trudności, ponieważ ich koniugowane zasady są dobrymi nukleofilami i są nawet słabszymi zasadami niż alkohole. Poniższe równania ilustrują niektóre reakcje substytucyjne alkoholi, na które mogą mieć wpływ te kwasy. Podobnie jak w przypadku halogenków alkilowych, substytucja nukleofilowa 1º-alkoholi przebiega za pomocą mechanizmu SN2, podczas gdy 3º-alkohole reagują za pomocą mechanizmu SN1. Reakcje 2º-alkoholi mogą zachodzić za pomocą obu mechanizmów i często wytwarzają pewne przestawione produkty., Liczby w nawiasach obok wzorów kwasów mineralnych przedstawiają procent wagowy stężonego roztworu wodnego, w postaci, w której te kwasy są zwykle używane.
chociaż reakcje te są czasami określane jako „katalizowane kwasem”, nie jest to ściśle poprawne. W ogólnej transformacji silny kwas HX jest przekształcany w wodę, bardzo słaby kwas, więc co najmniej stechiometryczna ilość HX jest wymagana do całkowitej konwersji alkoholu na halogenek alkilowy., Konieczność stosowania równoważnych ilości bardzo mocnych kwasów w tej reakcji ogranicza jej przydatność do prostych alkoholi typu pokazanego powyżej. Alkohole z grupami wrażliwymi na kwasy oczywiście nie tolerują takiego traktowania. Niemniej jednak pomysł modyfikacji grupy funkcyjnej-OH w celu poprawy jej stabilności jako anionu odchodzącego może być realizowany w innych kierunkach. Poniższy schemat pokazuje niektóre modyfikacje, które okazały się skuteczne. W każdym przypadku grupa hydroksylowa jest przekształcana w ester mocnego kwasu. Pierwsze dwa przykłady pokazują estry sulfonianów opisane wcześniej., Trzeci i czwarty przykład pokazują tworzenie estru fosforynowego (X reprezentuje Pozostałe brominy lub dodatkowe podstawniki alkoholowe) i estru chlorosulfinowego, odpowiednio. Wszystkie te grupy opuszczające (w Kolorze Niebieskim) mają sprzężone kwasy, które są znacznie silniejsze niż woda (o 13 do 16 Mocy dziesięciu); zatem anion opuszczający jest odpowiednio bardziej stabilny niż jon wodorotlenkowy. Związki mezylowe i tosylowe są szczególnie użyteczne, ponieważ mogą być stosowane w reakcjach substytucyjnych z szeroką gamą nukleofilów., Półprodukty wytwarzane w reakcjach alkoholi z tribromkiem fosforu i chlorkiem tionylu (dwa ostatnie przykłady) są rzadko izolowane, a reakcje te nadal wytwarzają bromek alkilowy i produkty chlorkowe.
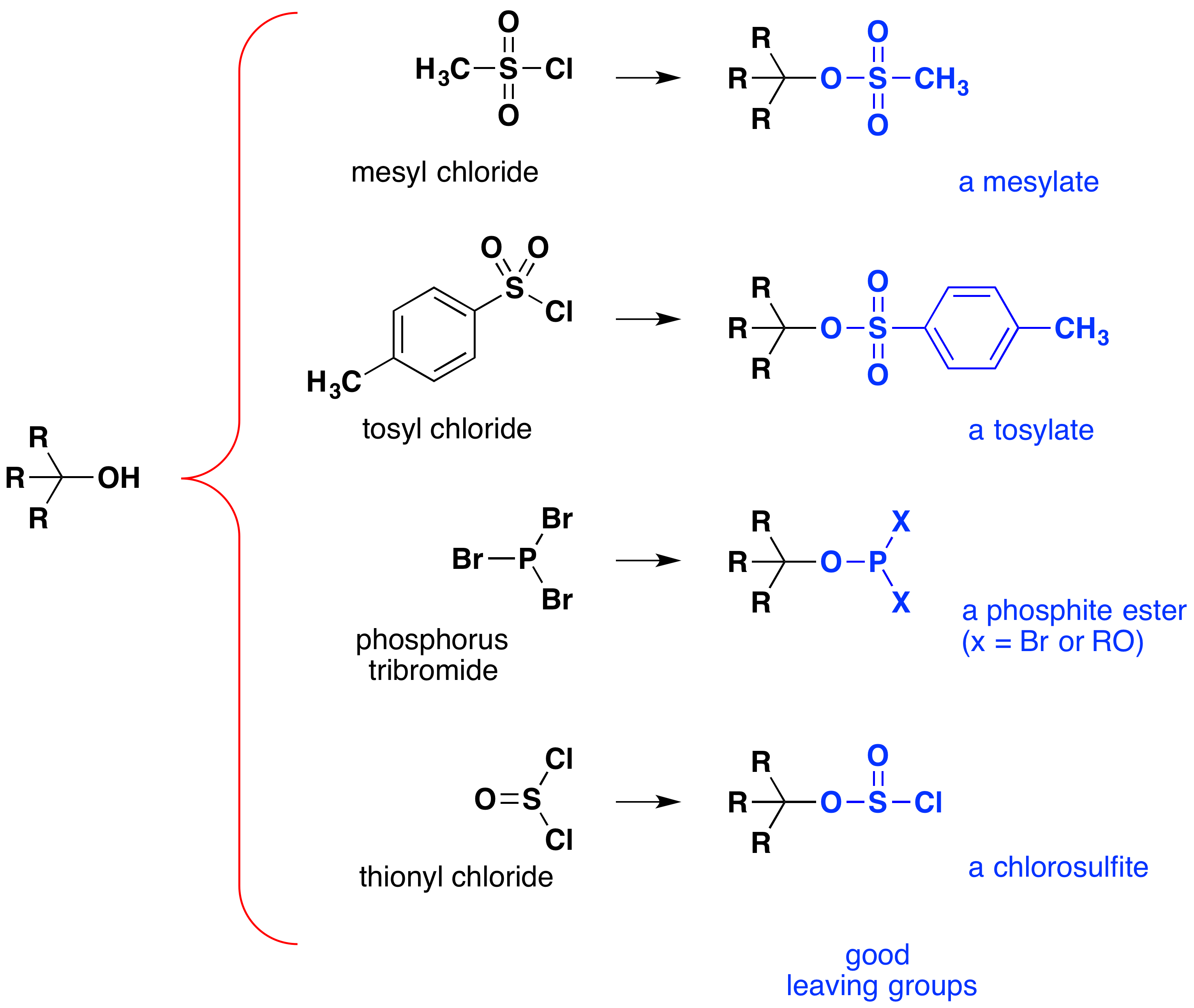
znaczenie półproduktów estrowych sulfonianów w ogólnych reakcjach substytucji nukleofilowej alkoholi można zilustrować następującą konwersją 1-butanolu do pentanenitrylu (cyjanku butylu), reakcji, która nie zachodzi z samym alkoholem., Z drugiej strony halogenki fosforu i tionylu działają tylko w celu przekształcenia alkoholi w odpowiednie halogenki alkilowe.,
| CH3CH2CH2CH2–OH + CH3SO2Cl | pyridine
|
CH3CH2CH2CH2–OSO2CH3 | Na(+) CN(–)
|
CH3CH2CH2CH2–CN + CH3SO2O(–) Na(+) |
Some examples of alcohol substitution reactions using this approach to activating the hydroxyl group are shown in the following diagram., Pierwsze dwa przypadki służą wzmocnieniu faktu, że sulfonianowe estrowe pochodne alkoholi mogą zastępować halogenki alkilowe w różnych reakcjach SN2. Kolejne dwa przypadki pokazują zastosowanie tribromidu fosforu w przekształcaniu alkoholi w bromki. Odczynnik ten może być stosowany bez dodatku zasady (np. pirydyny), ponieważ produkt kwasu fosforowego jest słabszym kwasem niż HBr. Tribromid fosforu najlepiej stosować z 1º-alkoholami, ponieważ 2º-alkohole często dają produkty uboczne przegrupowania wynikające z konkurencyjnych reakcji SN1., Należy zauważyć, że tlen eteru w reakcji 4 nie ma wpływu na ten odczynnik, podczas gdy alternatywna synteza przy użyciu skoncentrowanego HBr rozszczepia etery. Trichlorek fosforu (PCl3) przekształca alkohole w chlorki alkilowe w podobny sposób, ale chlorek tionylu jest zwykle preferowany do tej transformacji, ponieważ produktami nieorganicznymi są gazy (SO2 & HCl). Trijodek fosforu nie jest stabilny, ale może być wytwarzany in situ z mieszaniny czerwonego fosforu i jodu i działa w celu przekształcenia alkoholi w jodki alkilowe., Ostatni przykład pokazuje reakcję chlorku tionylu z chiralnym alkoholem 2º. Obecność Zasady organicznej, takiej jak pirydyna, jest ważna, ponieważ zapewnia znaczne stężenie jonów chlorkowych potrzebnych do końcowej reakcji SN2 półproduktu chlorosufitowego., W przypadku braku Zasady chlorosufity rozkładają się po podgrzaniu, aby uzyskać oczekiwany chlorek alkilowy z zachowaniem konfiguracji
alkohole trzeciorzędowe nie są powszechnie stosowane do reakcji substytucyjnych typu omawianego tutaj, ponieważ ścieżki reakcji SN1 i E1 są dominujące i trudne do kontrolowania.

znaczenie estrów sulfonianów jako półproduktów w wielu reakcjach substytucyjnych nie może być zawyżone., Z takich reakcji korzysta rygorystyczny dowód inwersji konfiguracyjnej, która zachodzi w miejscu substytucji w reakcjach SN2. Przykład takiego dowodu pokazany jest poniżej. Skróty dla częściej stosowanych pochodnych sulfonylowych podano w poniższej tabeli.
| Grupa Sulfonylowa | CH3SO2– | ch3c6h4so2– | brc6h4so2– | cf3so2– | |
|---|---|---|---|---|---|
| nazwa & Skrót., | Mesyl lub Ms | Tosyl lub Ts | Brosyl lub Bs | Trifyl lub Tf |
dowód inwersji

aby uzyskać pełniejsze omówienie reakcji substytucji hydroksylowej i opis innych selektywnych metod tej transformacji, kliknij tutaj.
współpracownicy
- William Reusch, profesor Emeritus (Michigan State U.,), Virtual Textbook of Organic Chemistry