Myotonische Dystrophie (DM)
Laden Sie unser Myotonische Dystrophie (DM) Fact Sheet
Was ist myotonische Dystrophie (DM)?
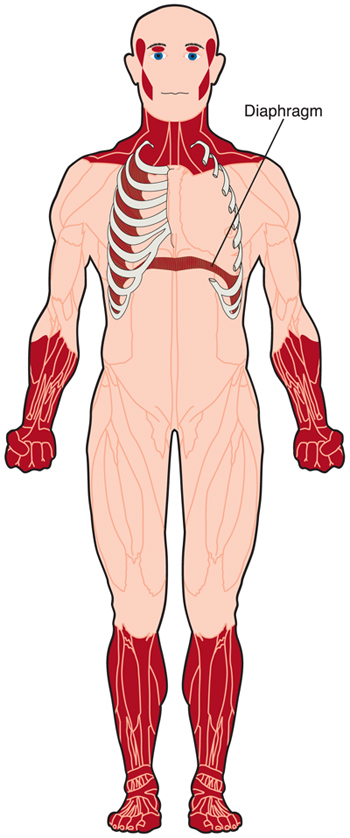 Myotonische Dystrophie (DM) ist eine Form der Muskeldystrophie, die Muskeln und viele andere Organe im Körper betrifft.
Myotonische Dystrophie (DM) ist eine Form der Muskeldystrophie, die Muskeln und viele andere Organe im Körper betrifft.
Das Wort “ myotonisch „ist die Adjektivform des Wortes“ Myotonie“, definiert als Unfähigkeit, Muskeln nach Belieben zu entspannen.
Der Begriff „Muskeldystrophie“ bedeutet progressive Muskeldegeneration mit Schwäche und Schrumpfung des Muskelgewebes.,
Myotonische Dystrophie wird oft als „DM“ in Bezug auf seinen griechischen Namen Dystrophia myotonica abgekürzt. Ein anderer Name, der gelegentlich für diese Störung verwendet wird, ist die Steinert-Krankheit, nach dem deutschen Arzt, der die Störung ursprünglich 1909 beschrieb.
Was verursacht DM?
DM ist in zwei Typen unterteilt.
Typ 1 DM (DM1), lange bekannt als Steinert-Krankheit, tritt auf, wenn ein Gen auf Chromosom 19 namens DMPK einen abnormal erweiterten Abschnitt enthält, der sich in der Nähe der Regulationsregion eines anderen Gens befindet, SIX5.,
Typ 2 DM (DM2), 1994 als mildere Version von DM1 anerkannt, wird durch einen abnormal erweiterten Abschnitt in einem Gen auf Chromosom 3 namens ZNF9 verursacht. DM2 wurde ursprünglich PROMM genannt, für proximale myotonische Myopathie, ein Begriff, der in Gebrauch geblieben ist, aber etwas seltener ist als der Begriff DM2.
Die erweiterten DNA-Abschnitte in diesen beiden Genen scheinen viele komplexe Auswirkungen auf verschiedene zelluläre Prozesse zu haben. Sowohl in DM1 als auch in DM2 wird die Wiederholungsexpansion in RNA transkribiert, bleibt aber im Protein unübersetzt.,
Diese Zustände sind einige der häufigsten Formen der Muskeldystrophie bei Erwachsenen. DM ist die häufigste Muskeldystrophie bei Erwachsenen europäischer Abstammung. Die Prävalenz von DM beträgt etwa 10 Fälle pro 100.000 Personen.1,2,3,4 Unter nicht-weißen Populationen ist DM1 ungewöhnlich oder selten.5,6,7,8 Berichte aus Europa deuten darauf hin, dass die Prävalenz von DM2 der von DM1 ähnelt.
Was sind die Symptome von DM?,
DM verursacht eine Schwäche der freiwilligen Muskeln, obwohl der Grad der Schwäche und die am stärksten betroffenen Muskeln je nach Art der DM und dem Alter der Person mit der Störung stark variieren.
Myotonie, die Unfähigkeit, Muskeln nach Belieben zu entspannen, ist ein weiteres Merkmal von DM. Zum Beispiel kann es für jemanden mit DM schwierig sein, jemandes Hand loszulassen, nachdem er sie geschüttelt hat.
Mit fortschreitender Krankheit kann das Herz einen abnormalen Rhythmus entwickeln und der Herzmuskel kann schwächer werden. Die zum Atmen verwendeten Muskeln können schwächer werden und insbesondere im Schlaf zu unzureichender Atmung führen.,9
Zusätzlich können bei Typ 1 DM die unwillkürlichen Muskeln, wie die des Gastrointestinaltrakts, betroffen sein. Schluckbeschwerden, Verstopfung und Gallensteine können auftreten.10,11 Bei Frauen können sich die Muskeln der Gebärmutter abnormal verhalten, was zu Komplikationen in Schwangerschaft und Wehen führt.12,13
Die Entwicklung von Katarakten (undurchsichtige Flecken in den Augenlinsen) relativ früh im Leben ist ein weiteres Merkmal von DM sowohl bei Typ 1 als auch bei Typ 2.,14
Die Gesamtintelligenz ist normalerweise bei Menschen mit DM normal, aber Lernbehinderungen und ein apathisches Verhalten sind in der Typ-1-Form üblich.15 Bei angeborenem DM1, das Kinder ab dem Zeitpunkt der Geburt betrifft, kann es zu schweren Beeinträchtigungen der kognitiven Funktion kommen. Diese Kinder können auch Probleme mit Sprach -, Hör -, Seh-und Sehstörungen haben.
Im Allgemeinen neigen die Symptome dazu, je früher DM1 beginnt. Weitere Informationen finden Sie unter Anzeichen und Symptome.
DM2 hat eine bessere Gesamtprognose als DM1. Die Symptome sind oft relativ mild und schreiten langsam voran., DM2 tritt selten in der Kindheit auf, und es ist keine angeborene Form von DM2 bekannt.
Was ist die Progression von DM?
Das Fortschreiten von DM variiert stark zwischen den Individuen, aber im Allgemeinen schreiten die Symptome allmählich voran. Die Lebenserwartung ist bei Patienten mit angeborenem DM1 deutlich reduziert und bei Patienten mit DM1 im Kindesalter und klassischem DM1 (Erwachsenenalter) wahrscheinlich reduziert.
Die häufigste Art von DM1-die Erwachsenenform — beginnt in der Adoleszenz oder im jungen Erwachsenenalter, oft mit Schwäche in den Muskeln von Gesicht, Hals, Fingern und Knöcheln., Die Schwäche ist langsam progressiv für diese und schließlich andere Muskeln.
Wenn DM1 früher im Leben als die Adoleszenz beginnt-die angeborenen und im Kindesalter auftretenden Formen der Krankheit -, kann es sich im Verlauf des Erwachsenenbeginns erheblich vom Typ des Erwachsenenbeginns unterscheiden. Kinder mit angeborenem DM1-Beginn zeigen, sobald sie die entscheidende Neugeborenenperiode der Atemmuskelschwäche mit Hilfe der assistierten Beatmung überleben, in der Regel Verbesserungen der motorischen und Atemfunktionen. Sie können kognitive Beeinträchtigungen, verzögerte Sprache, Schwierigkeiten beim Essen und Trinken und verschiedene andere Entwicklungsverzögerungen haben.,15
Die im Kindesalter auftretende Form von DM1 ist vor dem 10.Lebensjahr häufiger durch kognitive und Verhaltensstörungen als durch körperliche Behinderungen wie geistige Beeinträchtigung, Aufmerksamkeitsdefizite, Exekutivstörungen, Angstzustände und Stimmungsstörungen gekennzeichnet.17, 18, 19 Schließlich entwickeln sich Muskelsymptome in unterschiedlichem Maße.
DM2 ist im Allgemeinen eine mildere Krankheit als Typ-1. Es scheint keine angeborene Form zu haben und beginnt selten in der Kindheit.,
Im Gegensatz zu Typ 1 DM sind die Muskeln, die zuerst in DM2 betroffen sind, die proximalen Muskeln — diejenigen in der Nähe der Körpermitte — insbesondere um die Hüften herum. Einige Fingerschwächen können jedoch auch frühzeitig beobachtet werden. Die Störung schreitet langsam voran, aber die Beweglichkeit kann aufgrund der Schwäche der großen, tragenden Muskeln früh beeinträchtigt sein.
Wie ist der Stand der DM-Forschung?
Die Identifizierung der genetischen Mutationen, die DM1 und DM2 zugrunde liegen, und das Verständnis zumindest teilweise, wie die Mutationen Krankheiten verursachen, hat Wege für die Therapieentwicklung bei DM eröffnet.,
Die meisten Strategien, die derzeit entwickelt werden, zielen darauf ab, die schädlichen Auswirkungen der erweiterten DNA im DMPK-Gen (Typ 1) oder im ZNF9-Gen (Typ 2) zu blockieren. Weitere Informationen finden Sie unter Forschung, Im Fokus: Myotonische Dystrophie,und insbesondere DM Forschung: Suche nach Proteinen aus einem toxischen Netz zu befreien.’